摘要
将合成生物学和计算生物学的概念结合起来,可能会产生比现有药物更不容易产生耐药性的抗生素,而且还能对抗耐药感染。事实上,计算机引导策略与大规模并行高通量实验方法相结合,代表了抗生素发现的新范式。耐多药微生物引起的感染越来越致命,在当前的后抗生素时代,许多这些感染无法用我们现有的抗菌素库治疗。此外,我们可能已经用尽了自然界中产生的具有抗菌活性的大分子。耐药细菌的增加和缺乏新的抗生素类别显然需要开箱即用的策略。计算合成生物学的最新进展使抗菌剂的发展成为可能。新的分子描述符、遗传和模式识别算法是强大的工具,使我们离开发高效抗生素又近了一步。本文回顾了几种用于药物设计的计算工具和一些最近产生的候选抗生素,重点是基于肽的分子。设计策略可以产生多种合成抗菌肽,这可能有助于减轻耐药性的传播和对抗多重耐药微生物。
引言
这部分在介绍计算机辅助药物设计作为发现新药的潜在方法。由于组合合成和高通量筛选方法的进步,可以系统地修饰分子模板以发现药物。此外,尽管传统方法存在精度不高且产量低的问题,但近期技术进步已显著改善了化合物库的合成和功能筛选,使其更加高效和经济。面对多药抗性病原体引发的紧迫需求,当前缺乏真正的新型抗生素类别,因此亟需创新策略来应对抗药性问题。
计算机引导抗生素设计
详细阐述了利用计算机辅助设计新型抗生素的各种策略和方法,具体内容如下:
①新化合物的开发:通过对现有药物分子进行微调,例如轻微修改其功能基团或结合不同的化学片段,可以设计出新的活性化合物。这种方法使研究人员能够在已有的化合物基础上进行创新。
②数据集的重要性:计算机辅助药物设计通常需要输入一个包含丰富信息的数据集。这个数据集有助于筛选出最相关的药理特性,这些特性是定义化合物特定功能和效力的关键。例如,确定哪些药理属性需要优化,以提高化合物的生物活性。
③优化关键属性:确定需要优化的属性是设计新化合物的重要步骤。这些属性通常包括:毒性,评估化合物对人体细胞的潜在毒性,以确保新药的安全性;药代动力学特性,如吸收、分布、代谢和排泄(ADME),这些因素影响药物在体内的行为和疗效。
④设计方法分类:计算机辅助药物设计可以分为两种主要类型:基于结构的方法,利用已知的靶标结构,通过计算相互作用能量来优化分子的设计,这种方法通常能够提供更精准的设计方向;基于配体的方法,适用于缺乏结构信息的情况,通过已有的配体信息推测可能与目标结合的分子。
⑤抗生素的优化:计算机辅助设计技术尤其适用于肽类和蛋白质,因为这些生物分子具有多种功能特性。例如,计算机辅助设计可以帮助识别具有抗菌活性的肽类,以对抗多药耐药性微生物。
⑥效率与准确性:随着计算机技术的进步,药物发现的过程变得更加高效和经济。新的生物活性描述符和机器学习方法的应用,使得药物开发能够快速筛选出具有高效抗生素潜力的候选分子。
大型数据库作为输入,过滤最相关的药理学性质,定义这些化合物的特定功能和效力,生成的数据用于规划随后测试的一组新化合物,获得的结果用于分析新特性。重复这些步骤,直到可靠的评分功能导致优化的候选抗生素,然后根据其体内活性和毒性对其进行评估。该工艺产生的先导化合物有望用于临床试验。

多肽抗生素的计算导向探索
这部分详细探讨了计算机辅助方法在抗菌肽(AMPs)研发中的多种策略与应用。
肽的特性:AMPs是具有广泛生物活性的天然或合成分子,通常由20到50个氨基酸组成。它们在生物体内扮演着重要的角色,表现出抗菌、抗病毒、抗真菌和免疫调节等多种功能。由于其多样性和灵活性,抗菌肽被认为是新型抗生素的重要候选者。
计算算法的应用:当前有多种计算算法被开发用于描述肽的特性,包括机器学习和图论方法。这些算法旨在平衡效率和信息内容,能够快速处理大量数据,从而筛选出具有生物活性的肽。选择最优的描述符集取决于所需的生物功能预测和所采用的设计策略。
生物活性描述符:在开发抗菌肽的过程中,生物活性描述符起着关键作用,这些描述符包括:
- 可旋转键数和氢键受体/供体:影响肽的构象灵活性和结合能力。
- 表面积和芳香环含量:影响肽的疏水性和亲水性特征。
- 几何结构:影响肽与靶细菌的相互作用。
- 分子量:影响肽的细胞穿透能力和生物活性。
肽的优化:优化抗菌肽的过程通常使用分子建模和动态模拟方法,分析肽的结构-活性关系。通过这些方法,可以识别出哪些结构特征对抗菌活性至关重要,并指导肽的进一步设计。
先进的方法:计算方法的进步使得比较建模、预测模型和混合模型成为当前的研究热点。例如:
- 混合模型结合了多种算法的优点,提高了预测的准确性和可靠性。
- 预测模型使用机器学习技术从大量数据中识别重要的生物特征,并生成新的抗菌肽候选者。
- 比较建模通过对比已知的活性肽结构,推测新的肽的活性。
比较建模
比较建模(Comparative modeling)可以从复杂的自然结构(如蛋白质和酶)中识别出加密模板。通常情况下,大分子的蛋白水解过程会产生潜在的生物活性分子,但多数裂解片段并不一定具有生物活性。Pane等人描述了一种计算-实验框架,用于在验证人胃蛋白酶A激活肽的抗菌评分功能及其N端和C端片段具有AMPs活性后,发现新型的隐蔽抗菌肽。他们将来自胃蛋白酶原A3同工型的三种肽通过融合载体制备成重组形式,这种载体专门开发用于在大肠杆菌中表达潜在的毒性肽。由胃蛋白酶原A3衍生的重组肽被证明具有广谱抗菌作用,其最低抑菌浓度(MIC)范围为1.56至50 mmol/L,与完整的激活肽(1.56–12.5 mmol/L)对相同微生物的抑制效果相当。此外,该激活肽在pH 3.5的环境下对相关食源性病原体表现出杀菌活性,表明这种新类别的未被探索的抗菌肽可能有助于人体胃内的微生物监控。这些隐蔽肽对人类细胞无毒,但在体内表现出抗感染活性,在小鼠皮肤感染模型中将铜绿假单胞菌PAO1的细菌负荷降低了四个数量级。
预测模型
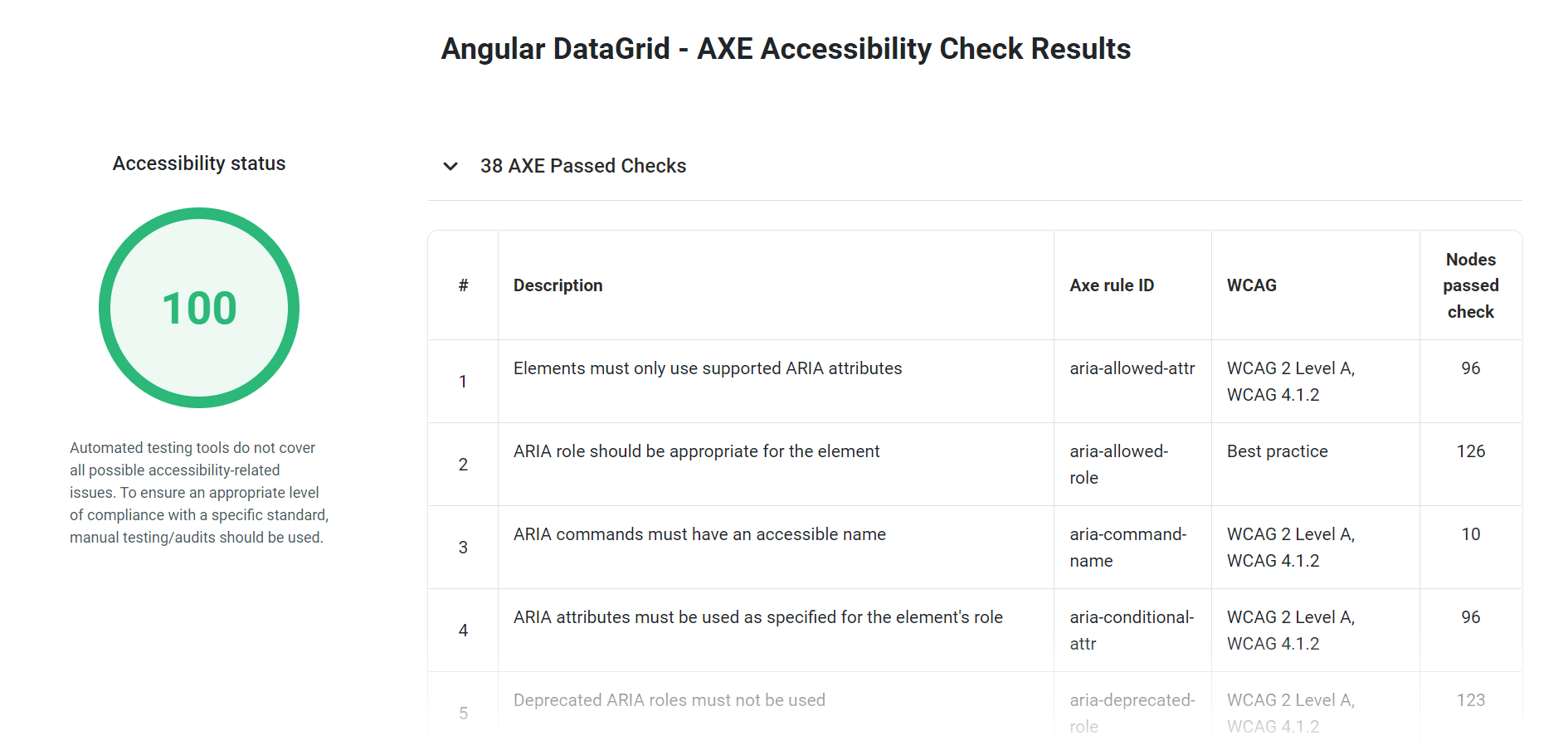
过去几十年中,已经提出了多种预测蛋白质和肽生物功能的方法。例如,有关蛋白质/肽折叠的众多研究在揭示生物物理、 生物化学和理化特性在结构-活性相关过程中的作用方面做出了重要贡献。理解这些特性将有助于设计定制的蛋白质和肽,并加深对蛋白质如何发挥其生物功能的理解。Huang等人详细介绍的从头蛋白设计技术展示了如何通过适当的理化原理和描述符来探索完整的序列空间,以帮助理解蛋白质的折叠机制。作者介绍了设计蛋白质的三种最相关的方法:结构预测、固定骨架设计和从头设计,并特别强调了从头蛋白设计。在结构预测中,序列是已知的,但骨架结构未知;在固定骨架蛋白设计中,序列未知但结构已知;而在从头设计中,序列和结构均未知。从头设计方法可能是一种设计新功能蛋白质和肽的替代途径,无需依赖天然模板的修改和优化。最近计算机科学的进展,如遗传算法、机器学习和深度学习,提供了其他实用工具,用于合理预测抗菌蛋白质和肽。
(a)模式识别算法是用于发现生物活性模板的重要工具,通过将其与已知的生物活性分子进行比较,这些模板被加密为天然生物分子。该技术使基于模板的新化合物的合理设计成为可能,而这通常是通过(b)分子建模和分子动力学进行的,这是分析结构-活性关系的非常有效的方法。(c)遗传算法也用于通过在适应度函数中排序的物理化学描述符从数据库生成候选抗生素。该功能对生成的新分子进行分类,并指出用于高通量筛选的先导化合物。(d)机器学习是应用于计算药物设计的最新技术之一。机器学习使用统计技术使计算机系统能够学习并逐步提高其性能,从而从物理化学和生物活性数据中生成生物活性化合物。

预测模型:遗传算法
遗传算法是设计AMPs的另一种方法,利用达尔文自然选择的基本原理来“进化”分子,使其达到所需的生物功能。该方法可以通过基于活性描述符和从AMP数据库中收集的信息构建适应度函数,对几乎所有新的AMP序列进行分类。尽管遗传算法生成的序列可能存在冗余,但该技术能够识别出具有独特成分和功能的新型人工AMPs。
Porto等人使用植物来源的模板,借助计算机辅助设计了番石榴富含甘氨酸的肽Pg-AMP1,并以此为模板,通过遗传算法生成了合成的guavanin肽。该算法通过适应度函数的描述性调整和在达到最优解之前的断点,生成了真正创新的肽,这些肽的设计来源于组合序列空间的进一步探索。设计中最有前景的类似物guavanin 2在低浓度下表现出杀菌作用,能通过诱导铜绿假单胞菌细胞膜的超极化破坏细菌细胞膜。
基于简单适应度函数的遗传算法还可以与其他设计模型结合,如Pane等人描述的模型,他们根据净电荷、疏水性和长度之间的线性关系预测了阳离子AMPs。这类研究中生成的输出数据集可以作为初始种群,通过遗传算法进行进化和生物优化。作者还指出,各理化参数对抗菌活性的相对贡献具有物种特异性。研究的一个重要发现是,一些细菌菌株对高电荷肽敏感,而另一些则对更加疏水的肽特别易感。
同样,分子结构,尤其是肽和蛋白质的构象分布,也可以通过算法来评估。Supady等人报告了使用遗传算法在构象空间中,搜索低能量构象。药物设计中分子结构的成熟原理方法用于评估从数据库中提取的氨基酸二肽构象数据集,并将系统搜索的性能与随机构象生成器的结果进行了对比。该算法准确地再现了参考数据,并且遗传算法对低能量构象空间的覆盖率显著优于作者选取的两种竞争方法,在相似的计算量下表现出更好的效果。
预测模型:机器学习
机器学习目前是计算机科学中最活跃的研究领域之一,广泛应用于生物工程和合成生物学等领域。Yoshida等人提出了一种概念验证方法,旨在有效优化AMPs的效能。具体而言,作者使用了一种闭环方法,将遗传算法、机器学习和体外评估结合在一起,以提高肽对大肠杆菌的抗菌活性。作者从一个小型阳离子天然模板中识别出了44种优选肽。经过三轮预测,这些优选肽的活性比野生型分子高出最多160倍。作者还成功地将无结构的肽转化为定义明确的螺旋分子,这些新肽的活性比野生型更强。该研究结果表明,机器学习能够提供工具,加速发现具有良好抗菌活性的AMPs,允许探索结构模式,并指示特定描述符如何影响生物活性。
基于机器学习的设计模型还可以用于创建具有特定作用机制的AMPs,Lee等人开发了一种支持向量机(SVM)分类器,用于研究对细菌膜有活性的α-螺旋AMPs的活性。该模型将功能的相似性与序列同源性相关联,作者利用SVM设计新型AMPs,该模型考虑了通过X射线散射确定的α-螺旋性以及体外抗菌活性。作者还观察到,他们的模型能够生成负的高斯膜曲率,作为抗菌活性的间接测量,这可能是一个非常有用的工具,因为它为许多AMPs共同的膜活性提供了拓扑基础。
预测模型:深度学习
深度学习,可能是当前可用的人工智能和计算机辅助设计技术中最有前景的一种。与遗传算法和机器学习中使用的理化或结构模式仅间接影响抗菌活性不同,深度学习生成的AMPs设计通常能直接预测AMP活性,这是一个显著的优势。然而,目前在这一领域内推进深度学习应用的研究相对较少。在这些研究中,神经网络的层次被分为多个层次,形成相关概念或决策树,每一层的输出数据会引导到更深层次的相关层,特别是从序列数据中衡量抗菌活性或结构的直接影响,同时还包括对理化特性的测量。
这种方法的一个缺点是需要大量可靠的生物数据作为自学习过程的输入,这对AMPs来说是一个挑战,因为目前缺乏肽测试的标准化程序,并且为避免预测误差所需的纯化肽合成成本较高。Veltri等人提出了一个具有少量层的简单深度神经网络,能够准确识别AMP。他们提出了一种用于识别肽序列中位置变化模式的模型,其精度高于目前生物信息学中常用的其他模型,如modlAMP、AntiBP2、CAMP和iAMPpred。Muller等人则提出了另一种用于计算指导的肽设计方法,即生成长短期记忆循环神经网络。利用该模型,作者能够提出组合式从头肽设计。该模型通过捕获α-螺旋AMP序列中的模式,并从学习的上下文中生成新的肽。作者报告说,预测为活性的AMPs中有82%确实表现出活性,而随机选择具有相同氨基酸分布的序列中这一比例仅为65%。这些策略为肽和蛋白质设计提供了替代方案,并消除了生成生物活性序列库所需的高强度高通量筛选的需求。
混合模型
上述模型的混合也生成了新的模型,这些模型往往表现出更高的准确性,因为它们能够捕捉每个组成模型的优点。Schneider等人提出的混合模型结合了网络模型和深度学习模型,该架构允许在自组织映射中使用多样化的多维描述符,将这些描述符转换为二维图像以进行进一步处理;这些二维图像被用作前馈神经网络的输入层。与缺乏自组织映射层的前馈网络分类器相比,这种模型具有更高的分类准确性和预测鲁棒性。
混合模型也被开发用于蛋白质设计和预测。Yan等人描述了一种旨在适当地将生物信息纳入从头对接(ab initio docking)的模型。作者开发了一种使用模板和非模板方法的混合对接模型。简而言之,当没有可用模板时,作者基于模板复合物或常规同源建模对单个组分的结构进行了建模。然后,通过传统的蛋白质-蛋白质对接预测该结构。利用这一方法,作者成功预测了16个CASP-CAPRI目标的正确模型,并对八个目标中的六个准确预测了蛋白质-肽的结合。
Gangopadhyay和Datta则采取了不同的方法,结合基于结构和基于配体的方法,识别出可配体位点,并使用识别出的抑制剂作为设计霍乱毒素活性抑制剂的参考配体。作者通过基于能量的方法识别了霍乱毒素的潜在配体结合位点,并将其发现与计算溶剂映射的结果进行了比较,从中识别出两个可能的配体结合位点,这些位点可能是针对霍乱的潜在靶点。类似的方法也可以用于基于结构的药物设计。
未来展望
在药物发现中使用的各种计算工具表明,并不存在一种根本优越的设计方法。不同方法的性能在很大程度上取决于所需的目标、可用数据和可用资源。评分函数的改进是计算辅助药物设计技术大规模应用的关键因素,这将涉及使用准确的描述符和精确的自由能测量。最有可能的是,结合基于结构和基于配体的计算方法将提供成功设计新型抗生素所需的准确性。
此外,随着复杂的结构和理化描述符的发展,计算设计方法变得越来越复杂——同时也变得更难以接近。因此,迫切需要将这些方法与新的分子动力学和分子建模进展结合起来。例如,Melo等人创建了一个集成的、综合的、可定制的、易于使用的套件,能够对靶蛋白或肽进行高质量分析,并生成精确的输出数据。通过结合量子力学和分子力学,Melo等人将两种广泛使用的分子动力学和可视化软件(NAMD和VMD)与量子化学软件包(ORCA和MOPAC)合并。作者展示了界面、设置、执行、可视化和分析可以成为各个专业水平的简便程序,并可应用于生成靶蛋白和肽的高质量分析。
寻找针对特定菌株的特异性的分子是预测模型的下一步,这些模型将在描述描述符与生物活性之间复杂关联方面发挥关键作用。Vishnepolsky等人最近描述了一种小型线性AMP的预测模型,该模型对特定革兰阴性菌具有抗菌活性。研究表明,可以通过结合半监督机器学习方法与基于密度的聚类算法,准确区分具有针对大肠杆菌ATCC 25922和铜绿假单胞菌ATCC 27853特定活性的肽,尤其是由18至27个残基组成的子集。Veltri等人还报告了一种基于序列模式构建和选择AMP的复杂描述符的方法。该方法通过识别肽的抗菌活性,并基于对革兰阳性菌、革兰阴性菌或两者的活性模型预测其靶向选择性。
结论
目前迫切需要设计真正新型的抗生素,同时提高发现和开发过程的准确性,并减少时间和成本。在可用于精确设计抗生素的方法中,计算工具和合成生物学工具最具潜力。当这些工具与组合合成和高通量筛选等实用方法相结合时,其价值更为显著。
肽设计不仅在生物应用方面提供了多种机会,也有助于理解稳定性、折叠机制及与其他分子相互作用背后的理化原理。目前,现有的评分函数偏向于疏水和静电相互作用,而忽视了氢键相互作用,这导致了毒性高、易聚集且缺乏选择性的序列。然而,计算辅助药物设计方法在准确的遗传算法引导下展示了良好的前景,成功生成了具有新颖序列、折叠方式、拓扑结构和功能的AMPs。随着新计算技术、更经济的计算资源和更准确的评分函数和优化算法的突破,进展正在快速加速。未来一定能够构建具有选择性、特异性和增强活性的计算设计肽类抗生素,用于治疗传染病。










![[笔记] Centos7 安装 Docker 和 Docker Compose 及 Docker 命令大全](https://i-blog.csdnimg.cn/direct/a2056f1cc67645c691741f33e56f434d.png)