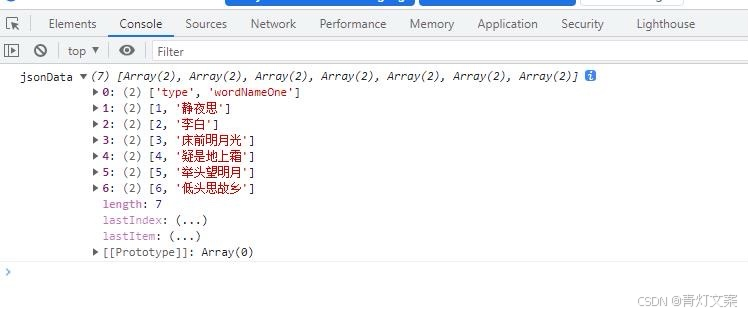
检查R语言安装包和依赖 .libPaths()

这里有一个简单的生物信息学分析案例,使用R语言处理基因表达数据。这个示例中,我们将导入模拟的基因表达数据,进行数据预处理(如归一化),并使用主成分分析(PCA)探索样本之间的关系。这个案例可以帮助了解常见的数据处理步骤,广泛用于基因组学、转录组学等分析。
案例:基因表达数据的主成分分析(PCA)
示例代码:
# 加载所需的R包
# 如果没有安装,需要先运行 install.packages("ggplot2") 和 install.packages("factoextra")
library(ggplot2)
library(factoextra)# 模拟基因表达数据(行表示基因,列表示样本)
set.seed(123) # 设置随机种子保证结果可复现
gene_data <- matrix(rnorm(1000), nrow = 100, ncol = 10)
rownames(gene_data) <- paste0("Gene", 1:100)
colnames(gene_data) <- paste0("Sample", 1:10)# 查看数据前几行
head(gene_data)# 数据标准化
gene_data_scaled <- scale(gene_data)# 进行PCA分析
pca_result <- prcomp(gene_data_scaled, center = TRUE, scale. = TRUE)# 查看PCA结果
summary(pca_result)# 绘制PCA图
pca_data <- as.data.frame(pca_result$x)
pca_data$Sample <- rownames(pca_data)
ggplot(pca_data, aes(x = PC1, y = PC2)) +geom_point(aes(color = Sample), size = 3) +labs(title = "PCA of Gene Expression Data", x = "PC1", y = "PC2") +theme_minimal()
代码解析:
- 加载包:
ggplot2用于绘图,factoextra提供PCA辅助功能。 - 生成数据:模拟了一个基因表达数据矩阵,100个基因和10个样本,使用正态分布生成随机数作为表达值。
- 标准化数据:将基因表达数据进行标准化,使每个基因的均值为0、标准差为1。
- 主成分分析:
prcomp()函数执行PCA分析,提取主成分。 - 绘制PCA图:用
ggplot2绘制主成分得分图,显示PC1和PC2,以观察样本间的分布。
注意事项:
在实际生物信息学分析中,数据可能来自真实的实验数据集,如RNA-Seq的表达矩阵。这时通常需要额外的数据清理、归一化和转化步骤。
![【题解】【排序】—— [NOIP2017 普及组] 图书管理员](https://i-blog.csdnimg.cn/direct/5d0c482919ed42469c5158761a9172df.jpeg#pic_center)