生信碱移
HiDDEN:抽丝剥茧
在具有病例和对照单细胞RNA测序研究中,样本级标签通常被直接赋予单个细胞,假设所有病例细胞都受影响。这种传统方法在受影响细胞比例较小或扰动强度较弱时,难以有效识别关键细胞及其标记基因(注意,这里以及后面所提到的扰动都是指不同分组下影响细胞的因素,比如病毒感染时病毒对细胞的转录影响)。同时,这些计算策略通常还假设:1) 分组标签能正确反映每个细胞的扰动状态;2) 分组标签所代表的扰动信号在潜在空间中占主导地位;3) 所有混淆变量均已知并可去除。但这些假设在细胞异质性和技术噪声较大的情况下往往难以满足,特别是当受影响细胞比例较小时。简单点说,传统方法假设所有来自疾病的细胞都受扰动,但实际上只有少部分细胞受影响,这种误差让研究者难以准确识别真正受影响的细胞及其特征。
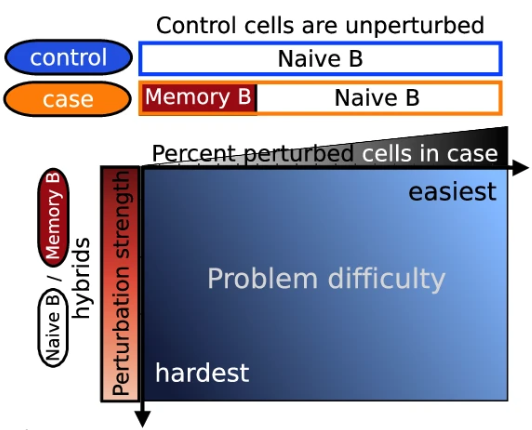
▲ 当前方法的问题。作者使用外周血单核细胞数据集中Naive B细胞和记忆 B 细胞的单细胞RNA-seq图谱进行了模拟。非记忆 B 细胞和记忆 B 细胞的表达谱相对相似,但在生物学上存在差异,因此适合模拟扰动引起的变化。作者构建了一个由Naive B(代表未扰动分组)细胞组成的对照样本和一个由Naive 和Memory B(代表扰动分组,Memory是受到扰动而改变的细胞)细胞组成的病例样本。但受扰动的关键细胞极少时,比如在病例条件下有5%的记忆B细胞会导致标准降维工作流程产生高度异质的降维潜在空间,从而无法检测到受干扰细胞的位置。沿两个轴的定义:病例样本中受扰动细胞的百分比(x 轴)和扰动强度(y 轴)。当受影响的细胞数量较少且受影响细胞与未受影响细胞之间的差异较小时,检测扰动最具挑战性。
为了解决这一问题,来自麻省理工学院-哈佛大学Broad研究所的研究人员提出了HiDDEN方法,于2024年11月2日发表于Nature Communications[IF:14.7]。该方法将样本级标签转化为细胞特异的连续扰动得分,进而根据扰动状态生成精细化的单个细胞标签。通过这种方式,HiDDEN显著提升了检测扰动相关基因的能力,有助于准确识别扰动标记并增强检测功效。在实验中,HiDDEN展示出能够克服传统方法的局限性,恢复出多个真实扰动信号,准确识别相关细胞亚群。

▲ DOI: 10.1038/s41467-024-53666-8
本文简要介绍HiDDEN的运行过程,更多细节可以查看原文或者下方链接:
-
https://github.com/tudaga/LabelCorrection/tree/main
0.环境配置与示例数据
环境配置:
HiDDEN基于python软件的scanpy单细胞流程,使用git clone克隆github项目仓后可以通过conda在环境中进行安装配置:
git clone https://github.com/tudaga/LabelCorrection.git
conda env create --file environment.yml
① 相关py库的导入:
import os
import sys
root_path = os.path.abspath('./..')
sys.path.insert(0, root_path )import itertools
import functools
from tqdm import tqdmimport pandas as pd
import numpy as np
import sklearn
import sklearn.linear_model
import sklearn.cluster
import sklearn.metrics
import matplotlib.pyplot as plt
import seaborn as snsimport hiddensc
from hiddensc import utils, files, visimport scanpy as sc
import scvi
import anndatautils.set_random_seed(utils.RANDOM_SEED)
utils.print_module_versions([sc, anndata, scvi, hiddensc])
vis.visual_settings()
② 加载并准备示例数据集
在示例教程中,作者使用10x Genomics免费提供的PBMCs数据集中Naive B细胞和记忆B细胞单细胞RNAseq数据的半模拟病例/对照单细胞数据集(如本文开头所描述的那样)。对照样本只包含Naive B细胞,病例样本由15%的记忆B细胞和85%的Naive B细胞组成(病例样本中15%的记忆B细胞可以理解为模拟的受到扰动因素干扰后产生的关键细胞亚群)。以该数据集为例,接下来展示如何识别病例样本中是否存在对照样细胞,并分离出真正受扰动的细胞亚群(这里是15%的记忆B细胞)。有关该数据集生成的更多详情,铁子们可以参阅原文。
REFIX = f'naiveB_1900_memoryB_154'
at_data_dir = functools.partial(os.path.join, root_path, files.DATA_DIR)
adata = sc.read(at_data_dir(f'{PREFIX}_raw.h5ad'))
adata.obs['binary_label'] = adata.obs['batch']=='Case'
hiddensc.datasets.preprocess_data(adata)
hiddensc.datasets.normalize_and_log(adata)
adata
#AnnData object with n_obs × n_vars = 2054 × 14603
# obs: 'barcodes', 'batch', 'perturbed', 'binary_label', 'n_genes'
# var: 'gene_ids', 'n_cells'
# uns: 'log1p'
# layers: 'counts'# Prepare directories
at_figure_dir = functools.partial(os.path.join, root_path, 'figures', 'tutorial')
os.makedirs(at_figure_dir(), exist_ok=True)
at_results_dir = functools.partial(os.path.join, root_path, files.RESULT_DIR, 'tutorial')
os.makedirs(at_results_dir(), exist_ok=True)
at_train_dir = functools.partial(os.path.join, root_path, files.RESULT_DIR, 'tutorial', 'training')
os.makedirs(at_train_dir(), exist_ok=True)print(f'Generating results at {at_results_dir()}')
# Generating results at /home/jupyter/LabelCorrection/results/tutorialhiddensc.datasets.augment_for_analysis(adata)
sc.pl.umap(adata, color=['batch'], s=120)
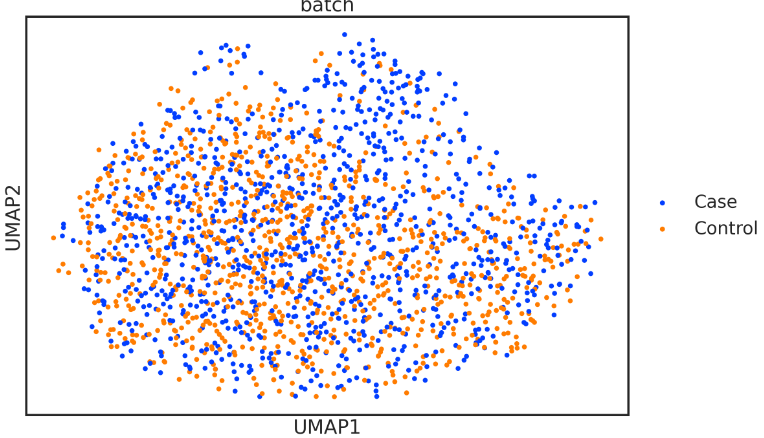
如图所示,无论是"病例 "还是 "对照 "的标签,都没有明显的富集群,此时标准分析工作流程无法识别出扰动的记忆B亚群。
1.HiDDEN分析
① 获得降维表示:
HiDDEN框架的第一步是计算单细胞数据的降维表示(表示是指高维基因表达降维后的潜空间,即PCA、UMAP、TSNE,不过最好选择能够良好表示细节的算法)。在这里,可以加载已经计算过的表示,例如去除批次效应或其他混杂因素后的表示。当然,hiddensc框架提供了多种降维方法供您选择,包括PCA和scvi。
第一种方案是使用PCA:
feats = []
# 计算主成分的最佳数量
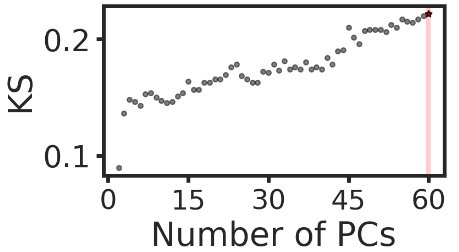
num_pcs, ks, ks_pval = hiddensc.models.determine_pcs_heuristic_ks(adata=adata, orig_label="binary_label", max_pcs=60)
optimal_num_pcs_ks = num_pcs[np.argmax(ks)]# 可视化
plt.figure(figsize=(4, 2))
plt.scatter(num_pcs, ks, s=10, c='k', alpha=0.5)
plt.axvline(x = optimal_num_pcs_ks, color = 'r', alpha=0.2)
plt.scatter(optimal_num_pcs_ks, ks[np.argmax(ks)], s=20, c='r', marker='*', edgecolors='k')
plt.xticks(np.append(np.arange(0, 60, 15), optimal_num_pcs_ks), fontsize=18)
plt.xlabel('Number of PCs')
plt.ylabel('KS')
plt.show()# 添加特征
x_pca = hiddensc.models.get_pca(adata, n_comps=optimal_num_pcs_ks)
feats['PCA'] = x_pca
第二种方案是使用SCVI或者LinearSCVI的模型嵌入:
feats = {}
# 指定嵌入模型的epoch训练轮数
n_epochs = 250
# 其实用到了两种模型嵌入
model_classes = [scvi.model.LinearSCVI, scvi.model.SCVI]
ks = [10]#, 20, 30, 40, 50]
combos = list(itertools.product(model_classes, ks))
# 训练与特征提取
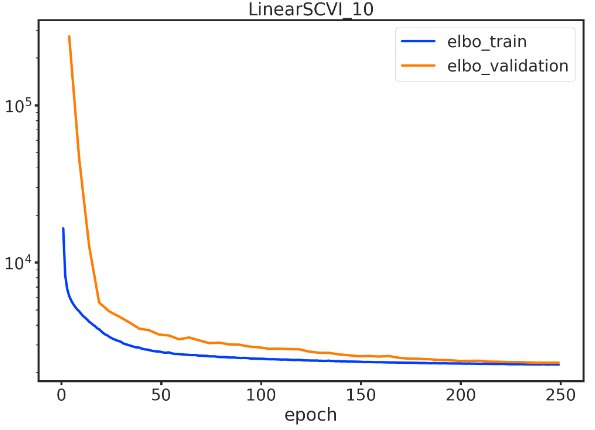
for model_cls, k in tqdm(combos):local_adata = adata.copy()name = f'{model_cls.__name__}_{k}'model_cls.setup_anndata(local_adata, layer="counts")model = model_cls(local_adata, n_latent=k)model.train(max_epochs=n_epochs, plan_kwargs={"lr": 5e-3}, check_val_every_n_epoch=5)train_elbo = model.history["elbo_train"][1:]test_elbo = model.history["elbo_validation"]ax = train_elbo.plot()test_elbo.plot(ax=ax)plt.yscale('log')#plt.savefig(at_train_dir(f'{name}.png'))plt.title(name)feats[name] = model.get_latent_representation()plt.show()del local_adata
值得注意的是,使用的降维方法全部放在一个字典feats即可。
② 生成预测
HiDDEN框架的第二个组成部分是一个预测模型,该模型根据降维特征和样本级标签进行训练(有意思的是,HiDDEN使用的是逻辑回归/SVM模型,还是比较容易实现的哈哈)。该模型的预测结果构成了每个细胞的连续扰动得分,可以用于探索扰动的潜在梯度效应,或进行聚类并生成精细的单个细胞二元标签。
feats = files.load_npz(at_results_dir('features.npz'))
y = (adata.obs['batch'].values == 'Case').astype(np.int32)
#y_true = (adata.obs['perturbed'].values == 'Memory B').astype(np.int32)
ids = adata.obs['barcodes'].values
pred_fns = {'logistic': hiddensc.models.logistic_predictions,'svm': hiddensc.models.svm_predictions}preds = [y]#, y_true]
info = [('batch', '','Case')]#, ('perturbed', '','Memory B')]
combos = list(itertools.product(feats.keys(), pred_fns.keys()))for feat_name, strat_name in tqdm(combos):rand_state=0x = feats[feat_name]p_hat, p_labels = pred_fns[strat_name](x, y, 1, rand_state)preds.append(p_hat)info.append((feat_name, strat_name, 'p_hat'))preds.append(p_labels)info.append((feat_name, strat_name, 'p_label'))cols = pd.MultiIndex.from_tuples(info)
pred_df = pd.DataFrame(np.array(preds).T, index=adata.obs['barcodes'], columns=cols)
pred_df.to_csv(at_results_dir('predictions.csv'))
pred_df
# batch PCA LinearSCVI_10 SCVI_10
#logistic svm logistic svm logistic svm
#Case p_hat p_label p_hat p_label p_hat p_label p_hat p_label p_hat p_label p_hat p_label
#barcodes
#naiveB_a_AAACCTGCACGGTAGA-1 1.000 0.543 0.000 0.546 1.000 0.437 0.000 0.461 0.000 0.375 0.000 0.401 0.000
#naiveB_a_AAACCTGCAGATGGGT-1 1.000 0.438 0.000 0.458 0.000 0.433 0.000 0.447 0.000 0.396 0.000 0.420 0.000
#naiveB_a_AAAGATGCATTTCAGG-1 0.000 0.302 0.000 0.439 0.000 0.359 0.000 0.409 0.000 0.370 0.000 0.409 0.000
#naiveB_a_AAAGCAAAGCCAACAG-1 0.000 0.393 0.000 0.449 0.000 0.407 0.000 0.430 0.000 0.469 0.000 0.481 0.000
#naiveB_a_AAAGCAAAGTGCCATT-1 1.000 0.531 0.000 0.500 0.000 0.562 0.000 0.535 0.000 0.418 0.000 0.441 0.000
#... ... ... ... ... ... ... ... ... ... ... ... ... ...
#memoryB_b_TGACTTTGTCACTTCC-1 1.000 0.629 1.000 0.545 1.000 0.563 0.000 0.545 0.000 0.548 1.000 0.534 1.000
#memoryB_b_TGTATTCGTTTGCATG-1 1.000 0.821 1.000 0.617 1.000 0.815 1.000 0.780 1.000 0.648 1.000 0.616 1.000
#memoryB_b_TGTATTCTCCTTTCTC-1 1.000 0.404 0.000 0.477 0.000 0.696 1.000 0.671 1.000 0.672 1.000 0.619 1.000
#memoryB_b_TTCTCCTCAGTCGATT-1 1.000 0.597 1.000 0.532 0.000 0.563 0.000 0.565 0.000 0.614 1.000 0.591 1.000
#memoryB_b_TTCTTAGTCGGCGCAT-1 1.000 0.762 1.000 0.587 1.000 0.761 1.000 0.716 1.000 0.679 1.000 0.625 1.000
#2054 rows × 13 columns
③ 可视化预测结果
根据上一小点的内容,分别设置降维以及预测模型的参数:
-
降维技术
DIM_RED:例如 "PCA"、"LinearSCVI_10"、"SCVI_10 -
预测模型
PRED_MODEL:如 "logistic"、"svm
IM_RED = 'PCA'
PRED_MODEL = 'logistic'
接下来,可以先可视化对照以及疾病组之间的p_hat差异(即所谓的二值化预测得分,模型输出的0-1范围概率)。
df = sc.get.obs_df(adata, ['batch', 'binary_label'])
df.columns = ['batch', 'binary_label']
df['p_hat'] = pred_df[f'{DIM_RED}'][f'{PRED_MODEL}']['p_hat'].values
df['new_label'] = pred_df[f'{DIM_RED}'][f'{PRED_MODEL}']['p_label'].values
df[' '] = 1
# 可视化
plt.figure(figsize=(6, 4))
sns.violinplot(data=df, x="batch", y="p_hat", linewidth=1, scale='count', cut=0, order=['Control', 'Case'])
plt.show()
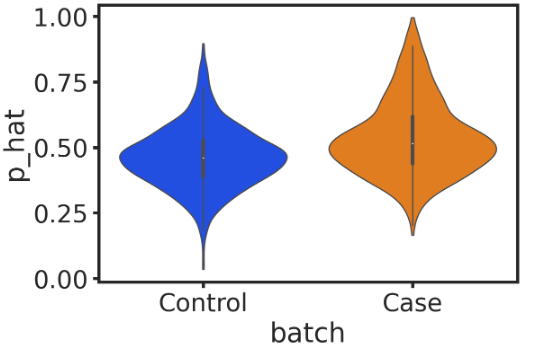
然后,可以将细胞分为三种:控制组中的B细胞(默认没有扰动)Control_L0、疾病组中的B细胞类型01Case_L0(没有扰动)、疾病组中的B细胞类型1Case_L1(受到扰动的)。可视化疾病样本中分离的关键扰动细胞群Case_L1:
# 设置标签
conditions = [(df['binary_label']==0),(df['binary_label']==1) & (df['new_label']==0),(df['binary_label']==1) & (df['new_label']==1)
]
values = ['Control_L0', 'Case_L0', 'Case_L1']
df['three_labels'] = np.select(conditions, values)# 可视化
plt.figure(figsize=(10, 5))
sns.violinplot(data=df, x="three_labels", y="p_hat", linewidth=1, scale='width', cut=0, order=['Control_L0', 'Case_L0', 'Case_L1'], palette=['lightblue', 'lightblue', 'orange'])
plt.ylabel('HiDDEN continuous score')
plt.xlabel("")
plt.show()# 看一下Case_L1的数量,攻击298个
np.sum(df['three_labels']=='Case_L1')
#298
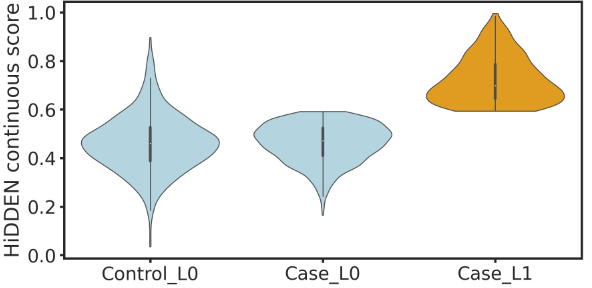
可以看到,Case_L1细胞群的二值化得分最高,是与疾病最潜在相关的细胞群体。
④ 这个时候就可以看看不同细胞群体的差异基因了!
adata.obs['new_label'] = pred_df[f'{DIM_RED}'][f'{PRED_MODEL}']['p_label'].values
de_genes = hiddensc.datasets.get_de_genes(adata, 'new_label')
de_genes.update(hiddensc.datasets.get_de_genes(adata, 'batch'))
de_genes
#{'0.0': array(['IGHD', 'TCL1A', 'BTG1', 'CXCR4', 'CD74', 'MEF2C', 'RPL18A',
# 'TMSB10', 'FCER2', 'HLA-DRA', 'MT-ND2', 'IGHM', 'HLA-DQB1',
# 'HLA-DRB1', 'IL4R', 'PLPP5', 'CD79B', 'HLA-DRB5', 'CD37', 'MT-CYB',
# 'RPS19', 'SERF2', 'MT-ATP6', 'LAPTM5', 'BIRC3', 'CD72', 'RPL21'],
# dtype='<U8'),
# '1.0': array(['B2M', 'COTL1', 'RPS18', 'HLA-B', 'EEF1A1', 'RP5-887A10.1',
# 'RPS14', 'GAPDH', 'HLA-C', 'SLC25A6', 'JCHAIN', 'IGHA1', 'NEAT1',
# 'S100A6', 'HSPA8', 'IGHG1', 'RPS15A', 'ITGB1', 'RPL14', 'TPT1',
# 'RPS29', 'CLECL1', 'S100A4', 'RPS20', 'HLA-A', 'VIM', 'PTP4A2',
# 'CD27', 'GPR183', 'RPS8', 'YWHAZ', 'EEF2', 'RPL27A', 'S100A10',
# 'LY6E', 'RPLP1', 'TOMM7', 'MALAT1', 'CRIP1', 'PRKCB', 'ACTG1',
# 'AL928768.3', 'RP5-1171I10.5', 'EVI2B'], dtype='<U13'),
# 'Case': array([], dtype='<U1'),
# 'Control': array([], dtype='<U1')}# 比较一下原始的分组标签以及现在的标签识别到的差异基因:
print(de_genes['Control'].shape)
print(de_genes['Case'].shape)
print(de_genes['0.0'].shape)
print(de_genes['1.0'].shape)
#(0,)
#(0,)
#(27,)
#(44,)
如上所示,使用原始样本级标签进行 DE 测试,没有发现 DE 基因,也完全无法识别 DE 基因。然而,使用经 HiDDEN 精炼的二进制标签进行 DE 测试,却能识别出一些 Naive B 和 Memory B 标记。当然,可以可视化看一下:
adata.obs['three_labels'] = df['three_labels']
dp = sc.pl.dotplot(adata=adata, use_raw=False,var_names= de_genes['1.0'][0:25], groupby='three_labels',categories_order=['Control_L0', 'Case_L0', 'Case_L1'],#standard_scale = 'var',title='Label 1 Top 25 DE genes',figsize=(10, 2))
#dp.add_totals().style(dot_edge_color='black', dot_edge_lw=0.5).show()
plt.show()
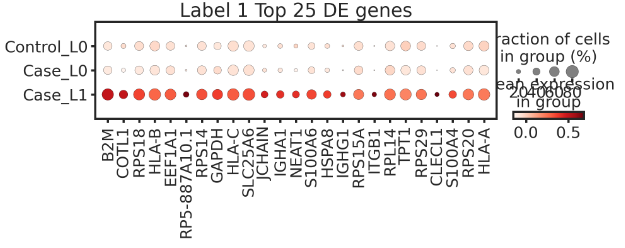
上面识别的Case_L1亚群表达多种Naive B细胞标记,验证了算法的准确性。
模型并不难理解
关键还是得有好点子
至于方法的准确性
大伙可以评估试试



















