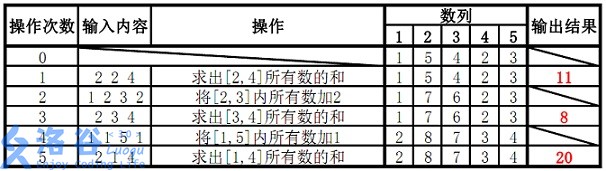
写在开头
大四期间,选修了一门智慧医疗的课程,期末考核为25分钟有关智慧医疗方面的汇报。一次偶然的课程汇报让我接触到了生物信息,也产生了浓厚的兴趣,同时加入了老师的研究生小组,开启了这段生物信息学习的旅途。至此开始学习并记录有关生物信息的基础知识。

1)前期软件的安装
问题1:为什么需要安装虚拟机呢?
对于生物信息分析来说,其分析主要分为两个部分,第一个部分分为上游分析,和下游分析;对于上游分析,一般数据量大,需要在服务器上运行分析;对于下游分析,即数据被服务器处理小后之后的数据,其实就可以被自己的个人笔记本电脑所运行分析了。对于上游分析来说,需要服务器,所以避免不了需要使用linux系统,为了学习上游分析相关的操作,就需要模拟出一台服务器,即就需要安装一个虚拟机。
1.1virtualBox的安装
Index of /jzjkkz_20210123/software/ 点击该网址
.dmg文件那个软件包是苹果系统的,.exe文件是windows操作系统的,选择自己的需要的下载即可。

安装的时候全程下一步就行。
配置虚拟机硬件信息
virtualBox下载好之后点开他,就会如下界面所示,刚下载完成后其实左侧界面是什么也没有的。

接下去其实模拟的就是选配一台服务器的过程,

这里建议为4096MB,太小不行,最少为4096。


然后一直默认的下一步直到下面这个界面,需要选择自己虚拟硬盘存储的位置,以及大小,这里选择128GB,然后点击创建。

到现在为止就等于我们以及买回来服务器,这个服务器现在还是一堆硬件,只有装上系统才能运行进行人机交互。
1.2 Ubuntu的安装
这里使用的Ubuntu版本为20.04.6桌面版本,因为本人其实刚学习使用linux,上来就装服务器版本的话害怕hold不住,所以就选择了桌面版本,实际的交互更加友好。
ubuntu 下载链接
左侧选择对应的版本,然后再点击DownLoad。

然后再点击下载即可。

下载好的镜像.iso文件放置在某个文件夹下。

给服务器装上系统
首先插上光驱,即选择刚刚下载好的.iso镜像文件的路径。网络选择桥接网络。

然后点击启动就可,然后具体的ubuntu的安装过程参考教程 ubuntu的安装
1.3 MobaXterm 的安装
这个软件,对于苹果用户来说就不需要安装而了;windwos电脑需要安装。

直接下载安装包解压就能用,不需要安装。直接双击就能直接使用

1.4 FileZilla 的安装

FileIlla的安装
1.5 MobaXterm和FileZilla连接服务器并使用
打开MobaXterm,点击Session。

然后查看服务器地址。

然后输入地址,用户名在点击ok。

连接成功,就可以输入用户密码从而操作服务器了。

打开FileZilla,同样的输入ip地址,用户名,密码,端口号写22。然后点击连接。


2)linux的相关命令
pwd命令 查看当前所在的路径

ls 查看当前路径下的所有文件夹

- ls -lh 的效果对比

cd 进入某个路径下使用cd

- cd . 效果:路径不变

- cd .. 效果:向前退了一步路径

cat 查看里面的内容

vim 是Linux中常用的编辑器

使用vim 加上文件的名称就可以打开该文件,打开后的文件如下所示,在这时,我们通过vim命令打开后,其实是进入了vim的一般模式,在一般模式中,其只能是进行光标的移动,通过小写的u进行撤销,小写的dd进行删除。

键盘英文状态下,按下i键就会进入vim的编辑模式,注意微小的区别,下方就会变成insert,此时就可以进行随意的编辑了,当编辑完成后,就可以按Esc键进行退出。

按下Esc键后,观察细微的变化,insert字样就会消失,但是好像还未退出,此时按住Shift+":"键,即输入:(英文状态下)。

然后输入wq(保存并退出),输入q!(强制不保存退出),输入q (不保存退出);然后再回车就可以真正退出了。

此时编辑的信息如下,然后现在关闭掉连接,即模拟电脑突然关机,那么此时编辑的信息是否会消失呢,是否会自动保存呢。我们来进行测试。

进行断开链接,模拟电脑突然关机。

意外在编辑的时候电脑中断退出,重新连接,再次输入vim README.md 进入该文件。

输入回车之后并没有如愿的再次进入文件,而是显示如下。

原因是电脑中断退出,系统其实自动的给我们保存相应的缓存文件README.md.swp ,此界面英文状态下点q即可退出。当我们使用 ls 命令查看文件的时候,发现好像根本没有README.md.swp文件。

其实只有输入ls -h -a ,就会如下图所示显示出来了。其实这个就是和我们windows中的隐藏文件一样,只是这个文件被隐藏了而已。只用通过ls -h -a才可以将隐藏的文件也显示出来。当然我们也是可以将某些文件设置为隐藏的, mv README.md .README.md 这个命令就可以将README.md文件隐藏了。

测试 mv README.md .README.md
将 README.md文件隐藏了。

输入r之后,显示如下,可以发现其实缓存文件不止一个,是在询问我们恢复到哪一个。

这里我们输入1,两次回车之后就显示如下

并没有因为突然的关机导致输入的数据丢失。
数据的打包和解压
使用zip或者gzip加文件名就可以将文件进行压缩。

解压使用unzip或gunzip加文件名就可以进行解压。

gzip和gunzip是linux自带的压缩和解压文件命令,要是想使用zip和unzip的话就需要安装这个两个命令的安装包。
将多个文件同时打包压缩

使用tar打包或者解包,-c表示打包,-f 后面接打包后的包的名称,然后加上所有要打包的文件。
tar -c -f all.tar 1.docx 2.docx.3.docx也可以使用如下命令,将所有的.docx文件进行打包了。
tar -c -f all.tar *.docx
同样使用tar进行解包,-x 表示进行解压,-f 后面跟着要解包的文件包。
tar -x -f all.tar结果如下。

打包压缩同时进行
tar -z -c -f all.tar.gz *.docx
解压解包同时完成
tar -z -x -v -f all.tar.gz
下载 FastQC软件,这里我新建了一个文件夹software来安装软件,使用wget来下载软件安装包。

然后解压
unzip fastqc_v0.12.1.zip
进入FastQC文件夹

运行fastqc程序,报错如下。

后面加上一个 -h 查看帮助文档,就可以看到该程序的简单用法。

配置环境变量,让我们的unzip 和zip等一样可以随时使用,而不用每次非得去找到该命令的绝对路径运行安装的命令。所以我们需要将fastqc命令的路径配置到环境变量中就可以随处运行。
输入echo $PATH后就可以直接的显示所有的环境变量路径。每个环境变量路径以冒号分隔开,

添加环境变量,使用export命令加上每个路径即可以将其添加至环境变量中,然后直接输入fastqc即可以执行该命令,而不再需要给出其前面的路径就可以直接找到其命令。只是由于我的虚拟机中并没有安装java,所以导致了其运行出错。

也可以使用以下命令将新的路径添加至环境变量,这样就可以免去将之前所有的路径再次复制过来。但是这些种添加环境变量的方式只对当前会话有效。
export PATH=$PATH:新添加的路径也可以使用which fastqc来查看查找到的fastqc命令的路径。

退出当前会话,再次进入后,再次使用fastac命令的时候,就会显示该命令找不到。

所以我们在添加环境变量的时候应添加到配置文件中,这样就可以不需要重复配置了。
vim 进入.bashrc,然后编辑,将路径添加至文档中,wq保存退出。


wq保存退出后,再次输入fastqc后,仍然显示找不到该命令,可是已经将路径添加进去了,但是为什么还是没有找到呢,是由于 .bashrc文件的原因,该文件只会在连接成功开机的时刻运行一次,所以使用命令exit退出后,再次进入后使用fastqc可以成功找到该命令。

也可以不退出,使用source .bashrc命令即可以重新运行,就可以将环境变量配置进去。

3)Anaconda的安装
可以使用wget下载anaconda,当然也是可以自己在windows上下载号linux版本的,然后再将其传送到liunx系统中。我是在windows上下载好再传送到linux的,因为我尝试使用wget下载,没有成功。

下载好后,就是一个.sh的文件,然后使用bash命令来执行该程序,然后会显示说按下enter键表示继续;然后就按下enter键,进入license。

一直按enter,直至最后,输入yes,回车。

确认安装路径,直接回车

询问是否需要初始化,输入yes。

然后就会安装成功

同样需要先将conda添加到环境变量中去,才能使用conda命令,在添加环境变量的时候,发现好像环境变量已经被添加了,其实是在提示我们时候需要初始化的时候,我们选择了yes,所以自动给我们加进去了,所以我们自己也就不用添加环境变量了,直接退出即可。

所以直接source .bashrc重新运行即可。会发现前面就会多出base的字符。输入conda后也可以正常的执行这个命令。

配置channels,按着如下输入vim .condarc。
![]()

配置好之后,保存退出即可,然后就可以开始使用conda安装相应的软件包了。

4)生物信息相关软件的安装
直接conda install 要安装的软件包 就可以安装对应的软件包了。

安装完成后,就可以使用which 查看安装的软件包在什么地方,

fastqc被直接安装在了anaconda bin目录下。这样使用conda安装软件的时候就可以直接使用conda命令安装,并且会自动的被安装在anaconda的bin目录下,而我们实际又将anaconda的bin的路径配置到了环境变量中,所以这样样我们就不需要再配安装好的软件的环境变量了。
然后可以将.condarc中的内容换成以下链接,使用清华的镜像,这样在之后安装软件的时候会更加的快速。输入vim .condarc,将之前写的内容更新为以下内容。
channels:- https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/pytorch/- https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/menpo/- https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/- https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/msys2/- https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/- defaults
show_channel_urls: true
至此我们是安装了两个fastqc了,那么我们输入fastqc这个命令,那到底运行的是哪个命令呢,是安装在/home/cce/anaconda3/bin/下,还是/home/cce/software/FsatQC/下的呢,其实我们通过命令echo $PATH查看,可以看见/home/cce/anaconda3/bin/是在环境变量前面的,所以实际输入命令fastqc是使用了/home/cce/anaconda3/bin/下面的命令。
已经装了两个个fastqc了,可以卸载掉与一个,这里卸载 /home/cce/software/FsatQC/下的。

继续安装blast、hisat2、trinity。

在安装剩余软件的时候,总是卡住,我尝试的很多方法都没有生效,换源,尝试了清华源,阿里源,以及北理工的源都以失败告终;又尝试了直接使用国外的安装,不使用镜像也以失败告终。

Collecting package metadata 失败显然就是获取资源的网站超时了,即源不对。
使用清华源下载,每一步几乎都被卡在了Solving Enviroment 这一步。然后感觉可能是由于base环境的原因,于是尝试创建一个新的环境供生物信息数据分析使用。

环境创建好后,激活环境,并进入该环境然后进行安装。


成功安装!